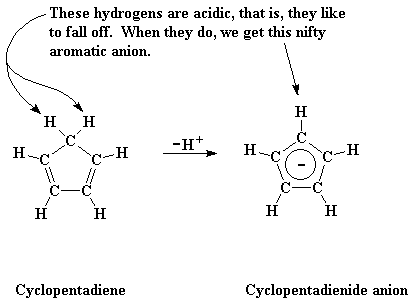
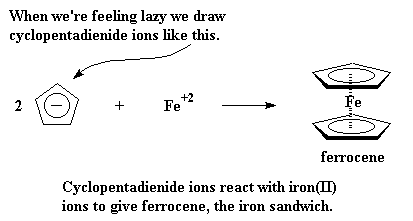
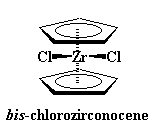
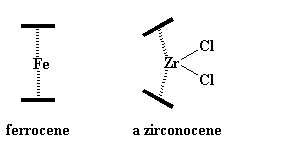
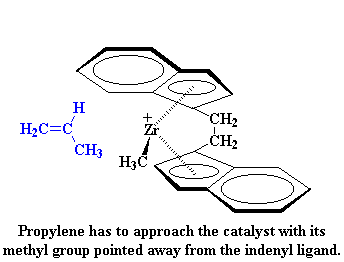
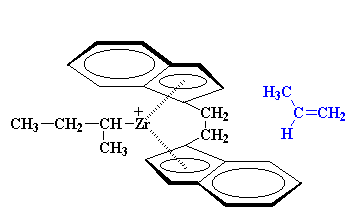
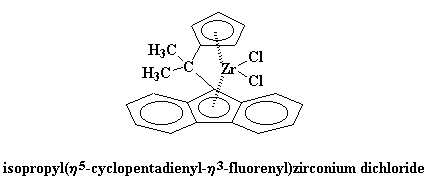
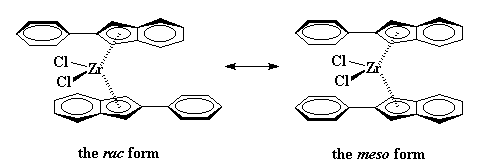
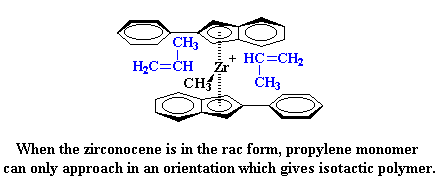
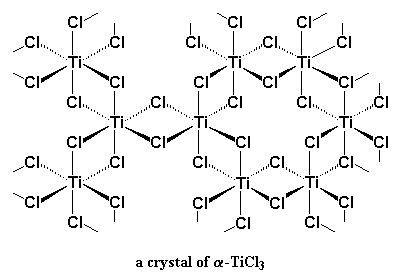
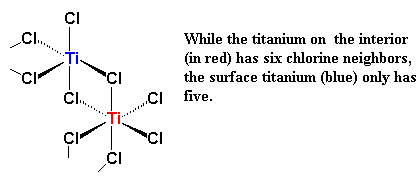
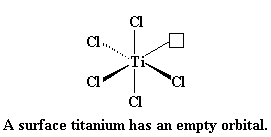
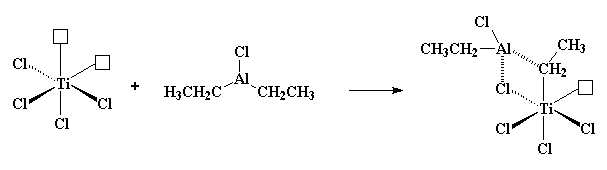
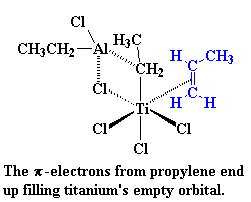
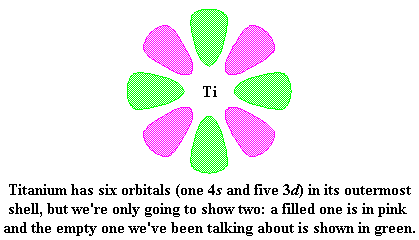
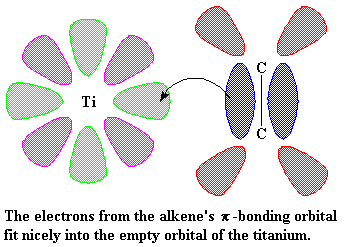
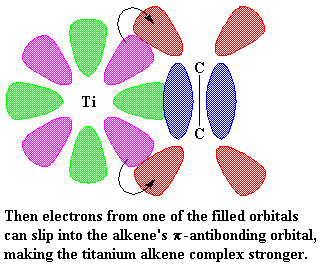
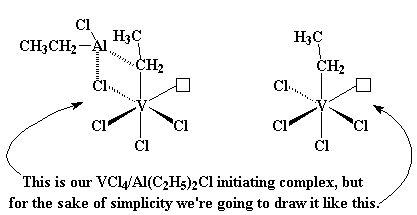
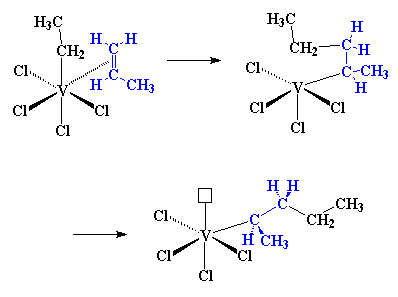
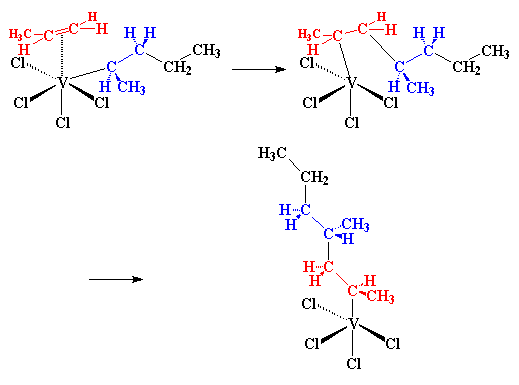
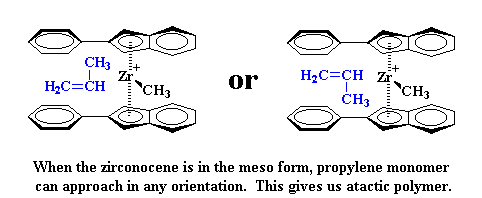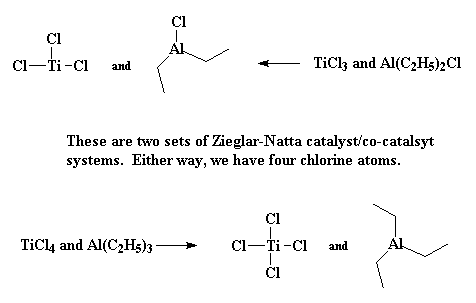|
Representative Work (2004-2012) |
Polymeric vesicles mimicking glycocalyx (PV-Gx) for studying carbohydrate-protein interactions in solution
Lu Su, Yu Zhao, Guosong Chen*, Ming Jiang |
|
Glycocalyx, the carbohydrate coat on cell surface, has been proved particularly important in a variety of biological events. In this work, polymeric vesicle mimicking glycocalyx (PV-Gx), as a simplified model system, is achieved via our NCCM strategy, using dynamic covalent bond between phenylboronic acid and sugar as the non-covalent linkage. Dynamic light scattering (DLS) has been employed to monitor the binding process between the sugars on PV-Gx and three different lectins. The results clearly proved that the PV-Gx constructed from the polymers with well-defined sugar units is a promising platform for the study of carbohydrate-protein interactions in solution without fluorescent labeling of proteins.

Su L, Zhao Y, Chen GS, Jiang M. Polym. Chem., 2012, DOI:10.1039/C2PY20110K. |
|
Self-assembly of particles—The regulatory role of particle flexibility
Kaka Zhang, Ming Jiang, Daoyong Chen* |
|
Aspects of the self-assembly of particles, which uses nanometer or micrometer sized building blocks to bridge the gap between microscopic and macroscopic scales, are reviewed. Particle self-assembly has been the focus of considerable research in recent years because it can lead to superstructures with a complexity inaccessible by molecular self-assembly, and functionalities entirely different from or superior to those of the primary particles. Examples in molecular self-assembly suggests that anisotropic interactions could be useful in promoting particle self-assembly, with the exception of colloidal crystallization, which requires particles of uniform size and shape. Anisotropic particles prepared by surface modification of precursor particles are often rigid and submicron or micron sized, and thus relatively strong isotropic van der Waals interactions tend to resist self-assembly into regular superstructures. In addition, the relatively large contact area between particles needed for a sufficient binding enthalpy to stabilize a superstructure is difficult for rigid spherical particles. In contrast, flexible anisotropic polymeric particles dispersed in solvents have been shown to self-assemble into various superstructures. The flexibility of primary anisotropic particles enables them to fuse and stabilize into a superstructure. Some flexible and multicomponent particles that are isotropic in common solvents can undergo deformation and sufficient material redistribution to anisotropically self-assemble into regular superstructures in selective solvents. The self-assembly is also driven by anisotropic interactions, which is induced during self-assembly rather than in the particles as synthesized. This review focuses on recent achievements in soft particle self-assembly and describes briefly the advancements in rigid particle self-assembly. The presentation is divided into discussion of self-assembly by the colloidal crystallization of isotropic rigid particles, anisotropic rigid particles, anisotropic soft particles and isotropic soft particles, in that order.

Zhang KK, Jiang M, Chen DY. Prog. Polym. Sci. 2011, DOI:10.1016/j.progpolymsci.2011.09.003 |
|
Does PNIPAM block really retard the micelle-to-vesicle transition of its copolymer?
Kongchang Wei, Lu Su, Guosong Chen*, Ming Jiang* |
|
Asymmetrically modified PNIPAM (Mw 10K), i.e. C12-PNIPAM-CA with a hydrophobic hydrocarbon chain -C12H25 (C12) at one end and a hydrophilic carboxyl group -COOH (CA) as the other, was prepared and found to form micelles with a core of the lightly associated hydrocarbon chains. When temperature is increased to the LCST of PNIPAM, the transformation from micelles to vesicles can be realized within 30 min, while the reverse process only takes a few minutes. Based on full monitoring of the transition process, it is proposed that the micelles serve as building blocks in constructing the vesicles via processes of combination, fusion, and etc., in which only local conformation adjustment of PNIPAM is involved.

Wei KC, Su L, Chen GS and Jiang M. Polymer, 2011, 52, 3647-3654. |
|
Cyclodextrin-based Inclusion Complexation Bridging Supramolecular Chemistry and Macromolecular Self-Assembly
Guosong Chen, Ming Jiang* |
|
In this invited review, we address how inclusion complexation has been employed and used to promote the recent developments in macromolecular self-assembly, especially with responsive functionalities. These include the amphiphilicity adjustment of macromolecules, non-covalent linkages for forming pseudo block copolymers and micelles, surface modification and functionalization of polymeric micelles and vesicles, and the combination of synthetic polymeric assemblies with biological moieties.

Chen GS, Jiang M*, Chem. Soc. Rev. 2011, 40, 2254-2266. |
Photoresponsive Pseudopolyrotaxane Hydrogels Based on Competition of Host–Guest Interactions
Xiaojuan Liao, Guosong Chen, Xiaoxia Liu, and Ming Jiang* |
|
A photo reversible hydrogel has been achieved via supramolecular competition. Briefly, a pseudopolyrotaxane Hydrogel formed by the inclusion complexation between α-cyclodextrin and poly(ethylene glycol) (PEG). After trans-azobenzene (trans-Azo) was added, the hydrogel transformed to solution because of the stronger binding ability of trans-Azo with α-CD than PEG. After UV light irradiation, trans-Azo isomerized to its cis form, which can not hold a-CD any more, which induced the formation of hydrogel again.

Liao XJ, Chen GS, Liu XX, Jiang M. Angew. Chem. Int. Ed. 2010, 49, 4409 –4413. |
Dual Stimuli-Responsive Supramolecular Hydrogel Constructed by Hybrid Inclusion Complex (HIC) formation
Jianghua Liu, Guosong Chen, Mingyu Guo, Ming Jiang |
|
A novel dual-responsive supramolecular hydrogel composed of an azobenzene (AZO) end- functionalized block copolymer PDMA-b-PNIPAM (AZO-(PDMA-b-PNIPAM)) and β-cyclodextrin-modified CdS quantum dot (β-CD@QD) has been demonstrated. Based on the host-guest inclusion complexation of AZO of the block copolymers and CD cavities on β-CD@QD, they form a hybrid inclusion complex (HIC). The inclusion complex and the domains of the collapsed PNIPAM chains serve as two distinct crosslinks and render the hydrogel excellent dual sensitivity to competitive hosts/guests substitution and to temperature variation.

Liu JH, Chen GS, Guo MY, Jiang M. Macromolecules. 2010, 43, 8086-8093. |
|
Non-covalently connected micelles (NCCMs): the origins and development ofa new concept
Mingyu Guo and Ming Jiang |
|
Nearly ten years ago, we suggested a new concept of non-covalently connected micelles (NCCMs) to describe and name a novel family of polymeric micelles. In NCCMs the components that form the core and shell are connected by hydrogen bonding instead of the normal covalent bonding that exists in all micelles formed from block copolymers. Investigations by us, as well as by others, in recent years have shown that the concept of NCCMs, and the methodology to attain them, are in fact much broader than our original suggestion.
Guo MY and Jiang M.Soft Matter 2009, 5, 495-500 |
|
Functional nanohybrids self-assembly from amphiphilic calix[6]biscrowns and noble metals
Bing Guan, Qing Liang, Yuan Zhu, Minghua Qiao, Jiong Zou and Ming Jiang |
|
One-dimensional nanohybrids made of organic templates of calixcrowns and in-situ formed metal nanoparticles were fabricated. The structure and catalytic properties of the hybrids were investigated.

Guan B, Liang Q, Jiang M, et al.JOURNAL OF MATERIALS CHEMISTRY 2009,19, 7610-7613 |
|
Dual Reversible Self-Assembly of PNIPAM-Based Amphiphilies Formed by Inclusion Complexation
Jiong Zou, Bing Guan, Xiaojuan Liao, Ming Jiang, and Fenggang Tao |
|
β-Cyclodextrin (β-CD)-ended linear poly(N-isopropylacrylamide) (β-CD-PNIPAM) and Frechet-type benzyl ether dendron with an azobenzene group (Gx-Azo) at the apex site form noncovalently connected amphiphiles (NCCAs) by inclusion complexation between the azo group and β-CD. The NCCAs self-assemble into vesicles in water. Optical switching of the assembly and disassembly is realized by alternating visible andUVirradiation, which causes the isomerization of the azo groups and their consequent complexation and decomplexation with β-CD. The structure and morphology of the vesicles were characterized by dynamic light scattering (DLS), static light scattering (SLS), SEM, TEM, and AFM. These photoresponsive vesicles can further respond to heat stimuli resulting in reversible aggregation and disaggregation of the vesicles.

Zou J, Guan B, Jiang M, et al. Macromolecules 2009, 42, 7465-7473 |
|
Self-assembly of amphiphilic calix[6] crowns: from vesicles to nanotubes
Guan, B(官冰); Jiang, M(江明); Yang, XG(杨晓刚), et al. |
|
In conclusion, to our knowledge, this communication presents the first e xample of a morphological transition from vesicles to nanotubes in self-assemblies of calixarenes. Unlike the remarkably broad and deep studies on self-assembly of block copolymers which started in the 1970’s, the investigation on self-assembly of the calixarenes is still at the very beginning. Great efforts are needed to exploit the relationship between the resultant morphologies and the structural parameters of the calixarenes, including the conformation of the framework, the structure and length of the attached hydrophilic chains etc, some of which is currently underway in our laboratory. xample of a morphological transition from vesicles to nanotubes in self-assemblies of calixarenes. Unlike the remarkably broad and deep studies on self-assembly of block copolymers which started in the 1970’s, the investigation on self-assembly of the calixarenes is still at the very beginning. Great efforts are needed to exploit the relationship between the resultant morphologies and the structural parameters of the calixarenes, including the conformation of the framework, the structure and length of the attached hydrophilic chains etc, some of which is currently underway in our laboratory. Guan, B; Jiang, M; Yang, XG, et al,SOFT MATTER 4(7)1393 |
Surface Modification of Polymeric Vesicles via Host-Guest Inclusion Complexation
Mingyu, Guo(郭明雨); Ming, Jiang(江明); Guangzhao, Zhang(张广照) |
|
As schematically summarized in Scheme 2, a novel kind of vesicle, which is reactive in supramolecular chemistry, was prepared through β-CD-ended polyether imide in water. On both the outer and inner surfaces of the vesicles, β-CD cavities are available for further surface modification via inclusion complexation between β-CD and adamantane-monoended PEG. For Ada-PEG2K and Ada-PEG1.1K, both the inner andou ter surfaces can be fully modified whereas for Ada-PEG5K the inner surfaces can be only partially modified. We emphasis that our surface modification of the vesicles is completely based on supramolecular chemistry and thus the guest polymers are no covalently connected to the surfaces. This study opens a new, simple, mild avenue to the surface modification and functionalization of the vesicles, which of course would promote their applications in various areas. ter surfaces can be fully modified whereas for Ada-PEG5K the inner surfaces can be only partially modified. We emphasis that our surface modification of the vesicles is completely based on supramolecular chemistry and thus the guest polymers are no covalently connected to the surfaces. This study opens a new, simple, mild avenue to the surface modification and functionalization of the vesicles, which of course would promote their applications in various areas.
Mingyu, Guo; Ming, Jiang; Guangzhao, Zhang,LANGMUIR 24(7) 10583 |
|
Polymer mortar assisted self-assembly of nanocrystalline polydiacetylene bricks showing reversible thermochromism
Gu, Y; Cao, WQ; Zhu, L, et al |
|
In summary, reversible thermochromism was achieved in hierarchically self-assembled PVP/PDA nanoaggregates using a hydrogen-bonding-assisted NCCM approach. Intr iguingly, a “bricks and mortar” structure in PVP/DA nanoaggregates was obtained by annealing the sample at a temperature (65°C) iguingly, a “bricks and mortar” structure in PVP/DA nanoaggregates was obtained by annealing the sample at a temperature (65°C)
slightly higher than the melting point of pure DA crystals (63°C). After topochemical polymerization, the resultant PVP/PDA “bricks and mortar” structurewas further stabilized at high temperatures (up to 120°C). Contrary to pure irreversible PDA crystals, PVP-intercalated PDA nanoaggregates showed reversible
thermochromic transitions in both aqueous suspension and dry films. The cooperative interaction between PVP and PDA through tethering carboxylic acid head groups in the side chains of PDA to PVP layers via hydrogen bonding is believed to be responsible for reversible conformational transitions between the “red” and “blue” states. This study not only opens a new avenue for further understanding of reversible thermochromism in polydiacetylenes but also leads to more effective ways of preparing thermally reversible polydiacetylene-based materials utilizing commercially available polymers and diacetylene monomers. Gu, Y; Cao, WQ; Zhu, L, et al, MACROMOLECULES 41(7)2299 |
|
Hydrogen-Bonded Dendronized Polymers and Their Self-Assembly in Solution
Xie D (谢荡), Jiang M (江明), Zhang GZ (张广照), Chen DY (陈道勇) |
|
Frechet-type benzyl ether dendrons of second and third generations with a carboxyl group (G2, G3) at the apex site could attach to poly(4-vinylpyridine) (PVP), forming hydrogen-bonded dendronized polymers (HB denpols) in their common solvent, chloroform.  The HB denpols show unique self-assembly behavior, forming vesicles in the common solvent under ultrasonic treatment.The structure and morphologyof the vesicles were characterized by dynamic light scattering (DLS), static light scattering(SLS), SEM, TEM,and AFM. The size of the vesicles decreases and thethickness of the vascular membrane increases as the molar ratio of Gx/PVP increases. The hydrogen bonding, pi-pi aromatic stacking of the dendrons, and the considerable difference in architecture between the dendron Gx and PVP are themain factors facilitating the assembly of the HB denpols in the common solvent. Xie D, Jiang M, Zhang GZ, et al. Chemistry-A European Journal 13 (12) 3346 |
| |
|
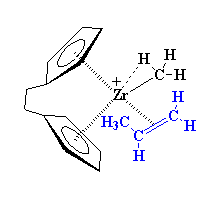
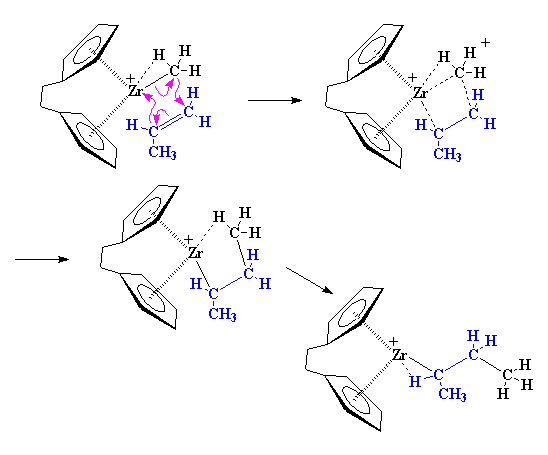
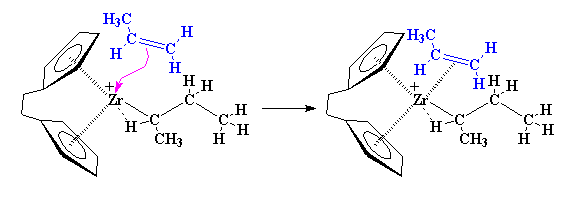
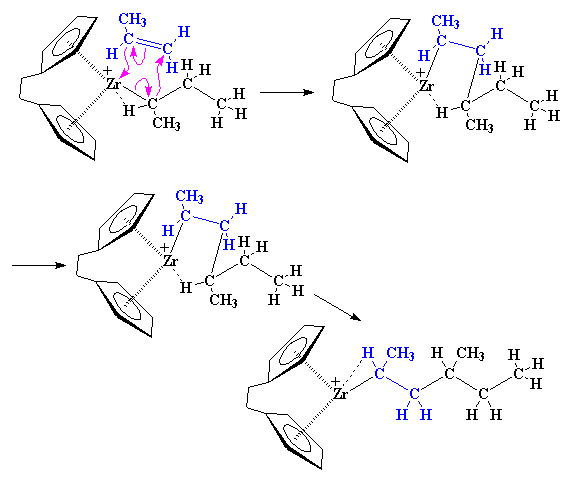
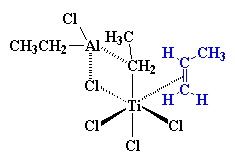
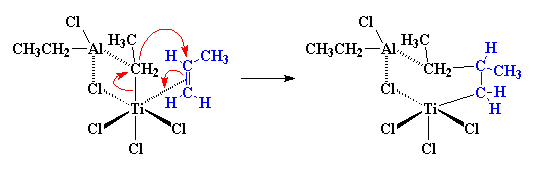
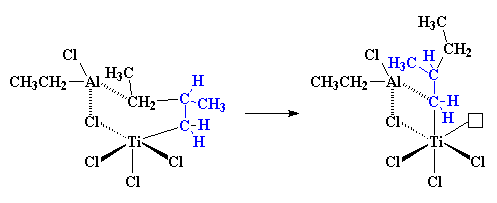
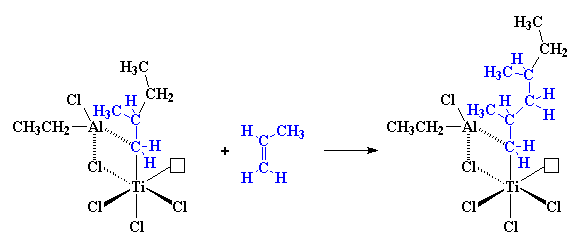
One-Pot Synthesis of Amphiphilic Polymeric Janus Particles and Their Self-Assembly into Supermicelles with a Narrow Size Distribution
Nie L (聂磊), Liu SY (刘世勇), Shen WM (沈文明), Chen DY (陈道勇), Jiang M (江明)
|
|
|
|
| |
|
Self-assembly of beta-casein and lysozyme
Pan, XY (潘晓贇); Yu, SY (俞绍勇); Yao, P (姚萍), et al. |
|
Abstract The self-assembly of β-casein and lysozyme, a linear and a globular protein with isoelectric point of pH 5.0 and 10.7, respectively, was studied. Polydisperse electrostatic complex micelles formed when mixing β-casein and lysozyme aqueous solutions. After the micelle solution was heated, lysozyme ge lated and β-casein was trapped in the gel, producing narrowly dispersed nanoparticles. The nanoparticles were characterized with laser light scattering, ζ-potential, steady state fluorescence, atomic force microscopy, and transmission electron microscopy. The nanoparticles have spherical shape and their sizes depend on the pH of the heat treatment and the molar ratio of β-casein to lysozyme. The nanoparticles display amphoteric property and are relatively hydrophobic at pH around 5 and around 10. The net charges on the surface stabilize the nanoparticles in the solution. lated and β-casein was trapped in the gel, producing narrowly dispersed nanoparticles. The nanoparticles were characterized with laser light scattering, ζ-potential, steady state fluorescence, atomic force microscopy, and transmission electron microscopy. The nanoparticles have spherical shape and their sizes depend on the pH of the heat treatment and the molar ratio of β-casein to lysozyme. The nanoparticles display amphoteric property and are relatively hydrophobic at pH around 5 and around 10. The net charges on the surface stabilize the nanoparticles in the solution.
Pan, XY; Yu, SY; Yao, P, et al. J. Colloid Interface Sci. 2007, 316 (2), 405
|
| |
|
Micellization induced by the inclusion complexation between β-CD and adamantly group (ADA)
Jing Wang (王竞), Ming Jiang* (江明) |
|
We developed a new route to fabricate non-covalently connected micelles (NCCM) of PGMA-CD/PtBA-ADA in aqueous media based on host-guest intera ction of β-CD and ADA. The presence of the β-CD cavities in the micellar shell provides broad opportunities to modify the micellar surface to meet different requirements in applications. Via shell-crosslinking and core-removal of the micelles, hollow spheres composed of β-CD-containing polymers were also obtained. These follow spheres possess multi-scale holes i.e. the large central one in size of 102 nm and many small β-CD cavities in 0.7 nm. ction of β-CD and ADA. The presence of the β-CD cavities in the micellar shell provides broad opportunities to modify the micellar surface to meet different requirements in applications. Via shell-crosslinking and core-removal of the micelles, hollow spheres composed of β-CD-containing polymers were also obtained. These follow spheres possess multi-scale holes i.e. the large central one in size of 102 nm and many small β-CD cavities in 0.7 nm.
Wang J, Jiang M J. Am. Chem. Soc. 2006, 128, 3703
|
| |
|
pH-Dependent Self-Assembly: Micellization and Micelle–Hollow-Sphere Transition of Cellulose-Based Copolymers
Hongjing Dou (窦红静), Ming Jiang* (江明) |
|
|
|
| |
|
A Novel Route to Thermo-Sensitive Polymeric Core-shell Aggregates and Hollow Spheres in Aqueous Media
Youwei Zhang (张幼维), Ming Jiang* (江明) |
|
The Poly(ε-carprolactone)(PCL)/Poly(N-isopropylacrylamide)(PNIPAM) core-shell particles was obtained by localizing the polymerization of NIPAM and crosslinker  methylene bisacrylamide around the surface of the PCL nanoparticles. The resultant particles were converted to hollow spheres by simply degradating the PCL core with enzyme. The attained hollow spheres is thermo-sensitive and displays reversible swelling and de-swelling around 32℃. Youwei Zhang, Jiang Ming et.al. Adv.Funct.Mater. 2005,15,695 |
| |
|
Optical Switching of Self-Assembly: Micellization and Micelle–Hollow Sphere Transition of Hydrogen-Bonded Polymers
Xikui Liu (刘习奎), Ming Jiang*(江明) |
|
The work reports reversible optical switching of micellization and micelle-hollow sphere transition in a blend solution of poly(4-phenylazomaleinanil- co-4-vinyl pyridine) (AzoMI-VPy) and carboxyl-ended polybutadiene CPB. Under UV irradiation, the trans-azobenzene units transformed into polar cis conformation and made AzoMI-VPy insoluble. Thus micelles formed with (AzoMI-VPy) core stabilized CPB shell through interpolymer hydrogen bonding. Upon visible light irradiation, the micelles quickly disassociated due to the cis form returning to  trans form. After core crosslinking, the micelles showed a reversible morphology change responding to light irradiation: visible light caused the formation of hollow spheres due to the core dissociation as a result of cis azobenzene turning to trans while UV light made the hollow spheres return to micelles due to the isomerization in the opposite direction. Xikui Liu, Ming Jiang Angew.Chem.Int.Ed 2006, 45, 3846 |
|
|
Preparation of Core-Stabilized Polymeric Micelles with a Mixed Shell Formed by Two Incompatible Polymers
Taoran Hui (惠陶然), Daoyong Chen* (陈道勇), Ming Jiang |
|
The preparation of core-stabilized micelles with PEO/PS as th  e shell by directly cross-linking P2VP in PS-b-P2VP and PEO-b-P2VP mixture in DMF, which is the common solvent of the block copolymers, using 1,4-dibromobutane as the cross-linker. Although the PEO chains and the PS chains are strongly incompatible, the cross-linking of P2VP blocks connects both the PEO and the PS chains to a common core and enables the sufficient mixing of the unlike blocks in the shell. Taoran Hui, Daoyong Chen et.al. Macromolecule.2005,38,5834 |
| |
|
Short-life core-shell structured nano-aggregates formed by the self-assembly of PEO-b-PAA/ETC (1-(3-dimethylaminopropyl) -3-ethylcarbodiimide methiodide) and their stabilization
Chunfeng Gu (顾春锋), Daoyong Chen* (陈道勇), Ming Jiang |
|
|
 The self-association takes place in the aqueous solution of PEO-b-PAA/ETC at the early stage of the reaction between PAA and ETC. Due to the reaction cycle of ETC with PAA (as indicated in the scheme), the aggregates have a limited life in water. After staying unchanged for several days in the aqueous solutions, the aggregates dissociate and finally disappear 1 to 3 weeks after their formation. The self-association takes place in the aqueous solution of PEO-b-PAA/ETC at the early stage of the reaction between PAA and ETC. Due to the reaction cycle of ETC with PAA (as indicated in the scheme), the aggregates have a limited life in water. After staying unchanged for several days in the aqueous solutions, the aggregates dissociate and finally disappear 1 to 3 weeks after their formation.
Chunfeng Gu, Daoyong Chen et.al. Macromolecule.2004,37,1666
|
| |
|
Self-assembly Based on Biomacromolecules
Shaoyong Yu (喻绍勇), Xiaoyun Pan (潘晓赟), Ping Yao*, Ming Jiang |
|
A new method was developed to produce nanogels with oppositely charged protein pairs or protein-polysaccharide pairs, such as chitosan-ovalbumin and ovalbumin-lysozyme pairs. The na nogels have core-shell structure. The dispersibility, the size and the hydrophobicity / hydrophilicity of the nanogels are pH responsible. Left figure is an illustration of the charge change of ovalbumin-lysozyme nanogels at different pH.Casein-g-dextran copolymer was prepared through the Maillard reaction. The copolymers form micelles at the pI of casein and the micelles dissociate when pH is away from t nogels have core-shell structure. The dispersibility, the size and the hydrophobicity / hydrophilicity of the nanogels are pH responsible. Left figure is an illustration of the charge change of ovalbumin-lysozyme nanogels at different pH.Casein-g-dextran copolymer was prepared through the Maillard reaction. The copolymers form micelles at the pI of casein and the micelles dissociate when pH is away from t he pI. β-carotene can induce the copolymer micellization through hydrophobic interactions during the β-carotene encapsulation procedure. Yu SY, Yao P, Jiang M, et al. Biopolymers 2006, 82, 148 Pan XY, Yao P, Jiang M, et al. J. Colloid and Inteface 2007, 315, 456 he pI. β-carotene can induce the copolymer micellization through hydrophobic interactions during the β-carotene encapsulation procedure. Yu SY, Yao P, Jiang M, et al. Biopolymers 2006, 82, 148 Pan XY, Yao P, Jiang M, et al. J. Colloid and Inteface 2007, 315, 456 |
| |
|
Interactions of Apo Cytochrome c with Alternating Copolymers of Maleic Acid and Alkene
Li Liang(梁丽), Ping Yao*, Ming Jiang |
The interactions of apo cytochrome c (apo cyt c) with poly(isobutylene-alt-maleic acid) (PIMA) and poly(1-tetradecene-alt-maleic acid) (PTMA) lead to apo cyt c a conformational change from random coil to -helical structure. The -helix content is influenced by the copolymer concentration, the length of alkyl chain of the copolymers, and media pH. The interactions of PIMA or PTMA with apo cyt c at neutral and alkali pH destroy the hydrophobic aggregation of PTMA or apo cyt c and form new complex particles. apo cyt c a conformational change from random coil to -helical structure. The -helix content is influenced by the copolymer concentration, the length of alkyl chain of the copolymers, and media pH. The interactions of PIMA or PTMA with apo cyt c at neutral and alkali pH destroy the hydrophobic aggregation of PTMA or apo cyt c and form new complex particles.
Li Liang, Ping Yao, et.al. Langmuir.2005,21,10662 |
以上工作简介中,图片均可点击放大查看,已经发表的工作均可点链接下载。因为图片较多,所以打开网页速度会较慢,请耐心等待。
|
|
|
| |
|
江明、陈道勇、姚萍联合课题组代表作 |
|
这里我们选择10篇代表性论文作简单交流,由此可以对我组研究工作的过去和现状有一个初步的了解。请点击文章名下载,下载之前请先安装Adobe AcrobatReader。
1. Ming Jiang, Hankun Xie
Miscibility and Morphology in Copolymer/homopolymer Blends, Prog.Polym.Sci.1991,16,977
本组建立伊始,研究由均聚物/嵌段共聚物的相容性问题起步,积5-6年研究成果,提出此类共混物相容性的“共聚物链构造效应”(chain-architectural effect),本组成果及国际学术界在此领域于八十年代的进展均总结于此文中。
2. Ming Jiang, Mei Li, Maoliang Xiang, Hui Zhou
Interpolymer Complexation and Miscibility Enhancement by Hydrogen Bonding, Adv.Polym.Sci.1999,146,121
九十年代初我组工作重点在高分子间的特殊相互作用和相容性的关系方面,该研究导致了我们逐渐形成和提出了“不相容-相容-络合转变”的概念。在此思想指导下对可控氢键体系的高分子间的络合展开了系统性研究,本文就这方面研究成果进行了总结。
3. Guangzhao Zhang, Ming Jiang, Chi Wu et al.
Formation of Novel Polymeric Nanoparticles, Acc.Chem.Res.2001,34,249,
我组于1995年发现含少量离子基团的碳氢链聚合物可在水中形成稳定的surfactant-free nanoparticle,其后,我们与吴奇教授合作对此类纳米粒子的形成规律作了深入研究。有关成果总结在这篇评述中。
4. Mei Li, Ming Jiang, Yunxiang Zhang, Qiu Fang,
Fluorescence Studies of Hydrophobic Association of Fluorocarbon-modified Poly(N-isopropylacrylamide), Macromolecules,1997,30,470
九十年代中期我组与中科院有机所章云祥教授就改性水性聚合物hydrophobic association 的问题开展了研究,本文是这方面成果的代表作。文章提出,经碳氟链修饰的Pyrene可以用为表征碳氟链改性的水溶性高分子的“靶向探针”,此文迄今被引用了62次。我们建议的探针已被法国和日本几个实验室用于多种碳氟微区的研究。
5.Min Wang, Guangzhao Zhang, Daoyong Chen, Ming Jiang,
Noncovalently Connected Polymeric Micelles Based on a Homopolymer Pair in Solutions, Macromolecules,2001,34,7172
Min Wang, Ming Jiang, Fangnin Ning, Daoyong Chen, Shiyong Liu, Hongwei Duan
Block-copolymer-free Strategy for Preparing Micelles and Hollow Spheres: Self-assembly of Poly(4-vinylpyridine) and Modified polystyrene, Macromolecules,2002,35,5980
在新世纪,本组的工作重点由高分子络合物转向了大分子自组装,目标是寻求获得规则纳米结构的新途径。以上两篇文章就是通过高分子间的氢键作用获得核-壳间非共价键合胶束(NCCM)的代表性文章。利用核-壳间无共价键特性,我们还在壳交联后将核溶解,在我组第一次获得聚合物空心球。
6. Hongwei Duan, Daoyong Chen, Ming Jiang, et al.
Self-assembly of Unlike Homopolymers into Hollow Spheres in Nonselective Solvent, J.Am.Chem.Soc.2001, 123,12097
刚性链在高分子组装中有独特的作用,本文证实,刚性聚酰亚胺和聚乙烯基吡啶在两者间的氢键相互作用和刚性链的规则排列倾向的驱动下,在其共同溶剂中,自组装为纳米空心球。此项成果开拓了我组刚性链作为组装单元的多方面的研究。
7.Xiaoya Liu, Ming Jiang, et al.
Micelles and Hollow Nanospheres Based on Epsilon-caprolactone-containing Polymers in Aqueous Media, Angew.Chem.Int.Ed.2002,41,2950
聚己内酯(PCL)和带PCL支链的水溶性高分子在水中可自组装为纳米球,经水溶性高分子主链的交联和PCL的酶降解,我们得到了空心球,这是高分子自组装的又一新途径。
8. Hongjing Dou, Ming Jiang, Huisheng Peng, et al.
pH-dependent Self-assembly: Micellization and Micelle-hollow-sphere Transition of Cellulose-based Copolymers, Angew.Chem.Int.Ed.,2003,42,1516
利用羟乙基纤维素和聚丙烯酸的接枝共聚物中主链和支链间的络合和解络合,我们成功地实现了它们在水中的胶束化和胶束-空心球转变,这两个过程都受pH控制。这一组装体系具有水溶性,可降解性,生物相容性,环境敏感性等多项特征。
9.Daoyong Chen, Huisheng Peng, Ming Jiang
A Novel One-step Approach to Core-stabilized Nanoparticles at High Solid Contents, Macromolecules,2003,36,2576
嵌段共聚物的胶束化通常只能在低浓度下实现,这大大限制了它的应用。本文提出,嵌段共聚物在共同溶剂中加入一种嵌段的交联剂,通过交联反应诱导胶束化。由于“非交联嵌段的屏蔽效应”,交联反应可在高浓度下(~10%)进行,导致纳米核交联胶束的形成,而避免了整体交联。这一途径已被证明有普遍意义。
10. Jie Gong, Ping Yao, Hongwei Duan, Ming Jiang, Shaohua Gu, Lijuan Chunyu
Structural Transformation of Cytochrome C and Apo Cytochrome C Induced by Sulfonated Polystyrene, Biomacromolecules, 2003,4,1293
我组近年来开拓了合成高分子/生物大分子相互作用研究的新方向,目前着重于合成高分子形成的多种微环境对蛋白质分子折叠的影响。本文是这一研究方向在Biomacromolecules上发表的第一篇文章。
|